3. Jaccard coefficient (J)
3.1. Description
Calculate the Jaccard similarity coefficient between two sets of genomic regions.
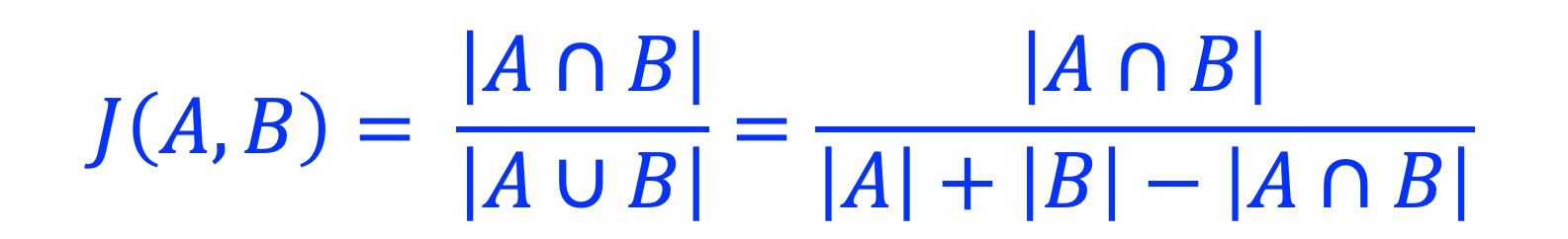
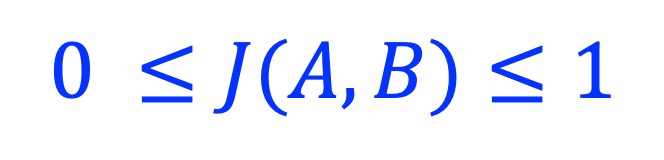
3.2. Usage
cobind.py jacard -h
usage: cobind.py jaccard [-h] [--nameA NAMEA] [--nameB NAMEB] [-n ITER]
[-f SUBSAMPLE] [-b BGSIZE] [-o] [-l log_file] [-d]
input_A.bed input_B.bed
positional arguments:
input_A.bed Genomic regions in BED, BED-like or bigBed format. The
BED-like format includes:'bed3', 'bed4', 'bed6',
'bed12', 'bedgraph', 'narrowpeak', 'broadpeak',
'gappedpeak'. BED and BED-like format can be plain
text, compressed (.gz, .z, .bz, .bz2, .bzip2) or
remote (http://, https://, ftp://) files. Do not
compress BigBed foramt. BigBed file can also be a
remote file.
input_B.bed Genomic regions in BED, BED-like or bigBed format. The
BED-like format includes:'bed3', 'bed4', 'bed6',
'bed12', 'bedgraph', 'narrowpeak', 'broadpeak',
'gappedpeak'. BED and BED-like format can be plain
text, compressed (.gz, .z, .bz, .bz2, .bzip2) or
remote (http://, https://, ftp://) files. Do not
compress BigBed foramt. BigBed file can also be a
remote file.
options:
-h, --help show this help message and exit
--nameA NAMEA Name to represent 1st set of genomic interval. If not
specified (None), the file name ("input_A.bed") will
be used.
--nameB NAMEB Name to represent the 2nd set of genomic interval. If
not specified (None), the file name ("input_B.bed")
will be used.
-n ITER, --ndraws ITER
Times of resampling to estimate confidence intervals.
Set to '0' to turn off resampling. For the resampling
process to work properly, overlapped intervals in each
bed file must be merged. (default: 20)
-f SUBSAMPLE, --fraction SUBSAMPLE
Resampling fraction. (default: 0.75)
-b BGSIZE, --background BGSIZE
The size of the cis-regulatory genomic regions. This
is about 1.4Gb For the human genome. (default:
1400000000)
-o, --save If set, will save peak-wise coefficients to files
("input_A_peakwise_scores.tsv" and
"input_B_peakwise_scores.tsv").
-l log_file, --log log_file
This file is used to save the log information. By
default, if no file is specified (None), the log
information will be printed to the screen.
-d, --debug Print detailed information for debugging.
3.3. Example
Calculate the overall Jaccard coefficient and peak-wise Jaccard coefficient between CTCF binding sites and RAD21 binding sites.
python3 ../bin/cobind.py jaccard CTCF_ENCFF660GHM.bed RAD21_ENCFF057JFH.bed --save
The overall Jaccard coefficient between CTCF_ENCFF660GHM.bed and RAD21_ENCFF057JFH.bed was printed to screen
2022-01-16 08:24:12 [INFO] Calculate Jaccard coefficient (overall) ...
A.name CTCF_ENCFF660GHM.bed
B.name RAD21_ENCFF057JFH.bed
A.interval_count 58684
B.interval_count 33373
A.size 12184840
B.size 11130268
A_or_B.size 18375623
A_and_B.size 4939485
Coef 0.2688
Coef(expected) 0.0042
Coef(95% CI) [0.2672,0.2713]
dtype: object
2022-01-16 08:24:40 [INFO] Calculate Jaccard coefficient (peakwise) ...
2022-01-16 08:24:40 [INFO] Read and union BED file: "CTCF_ENCFF660GHM.bed"
2022-01-16 08:24:40 [INFO] Unioned regions of "CTCF_ENCFF660GHM.bed" : 58584
2022-01-16 08:24:40 [INFO] Read and union BED file: "RAD21_ENCFF057JFH.bed"
2022-01-16 08:24:41 [INFO] Unioned regions of "RAD21_ENCFF057JFH.bed" : 31955
...
If --save was specified, the peakwise coefficients were saved to CTCF_ENCFF660GHM.bed_peakwise_scores.tsv and RAD21_ENCFF057JFH.bed_peakwise_scores.tsv, respectively.
$ head -5 CTCF_ENCFF660GHM.bed_peakwise_scores.tsv
chrom start end A.size B.size A∩B A∪B B.list Score
chr12 108043 108283 240 404 240 404 chr12:107919-108323 0.594059405940594
chr12 153232 153470 238 222 222 238 chr12:153236-153458 0.9327731092436975
chr12 177749 177989 240 NA NA NA NA NA
chr12 189165 189405 240 404 240 404 chr12:189072-189476 0.594059405940594
- column 1 to 3
The genomic coordinate of CTCF peak.
- column 4 (A.size)
The size of CTCF peak.
- column 5 (B.size)
The size (cardinality) of RAD21 peak(s) that were overlapped with this CTCF peak.
- column 6 (A∩B)
The size (cardinality) of intersection.
- column 7 (A∪B)
The size (cardinality) of union.
- column 8 (B.list)
List of RAD21 peak(s) that are overlapped with this peak. Multiple peaks will be separated by “,”.
- column 9 (Score)
The peakwise Jaccard coefficient.