8. Cooccurrence
8.1. Description
Use Fisher’s exact test to evaluate if two sets of genomic intervals (A and B) are significantly cooccured [1]. Genomic intervals (g) in the background BED file will be divided into 4 groups: a (A specific), b (B specific), c (A and B cooccur), and n (neith A nor B).
Not A |
A |
Total |
|
|---|---|---|---|
Not B |
n |
a |
n+a |
B |
b |
c |
b+c |
Total |
n+b |
a+c |
g = a + b + c + n |
Fisher’s exact test p-value is calculated as:
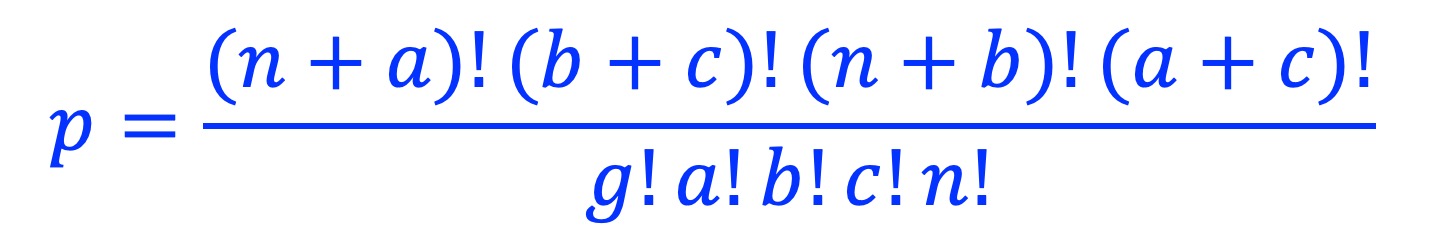
Odds ratio is calculated as:
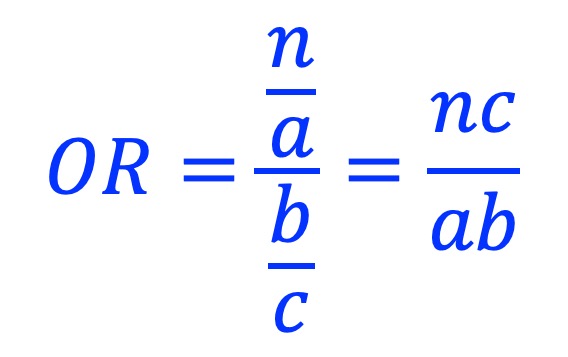
8.2. Usage
cobind.py cooccur -h
usage: cobind.py cooccur [-h] [--nameA NAMEA] [--nameB NAMEB] [--ncut N_CUT]
[--pcut P_CUT] [-l log_file] [-d]
input_A.bed input_B.bed background.bed output.tsv
positional arguments:
input_A.bed Genomic regions in BED, BED-like or bigBed format. The
BED-like format includes:'bed3', 'bed4', 'bed6',
'bed12', 'bedgraph', 'narrowpeak', 'broadpeak',
'gappedpeak'. BED and BED-like format can be plain
text, compressed (.gz, .z, .bz, .bz2, .bzip2) or
remote (http://, https://, ftp://) files. Do not
compress BigBed foramt. BigBed file can also be a
remote file.
input_B.bed Genomic regions in BED, BED-like or bigBed format. The
BED-like format includes:'bed3', 'bed4', 'bed6',
'bed12', 'bedgraph', 'narrowpeak', 'broadpeak',
'gappedpeak'. BED and BED-like format can be plain
text, compressed (.gz, .z, .bz, .bz2, .bzip2) or
remote (http://, https://, ftp://) files. Do not
compress BigBed foramt. BigBed file can also be a
remote file.
background.bed Genomic regions as the background (e.g., all
promoters, all enhancers).
output.tsv For each genomic region in the "background.bed" file,
add another column indicating if this region is
"input_A specific (i.e., A+B-)", "input_B specific
(i.e., A-B+)", "co-occur (i.e., A+B+)" or "neither
(i.e, A-B-)".
options:
-h, --help show this help message and exit
--nameA NAMEA Name to represent 1st set of genomic interval. If not
specified "A" will be used.
--nameB NAMEB Name to represent 2nd set of genomic interval. If not
specified "B" will be used.
--ncut N_CUT The minimum overlap size. (default: 1)
--pcut P_CUT The minimum overlap percentage. (default: 0.000000)
-l log_file, --log log_file
This file is used to save the log information. By
default, if no file is specified (None), the log
information will be printed to the screen.
-d, --debug Print detailed information for debugging.
8.3. Example
cobind.py cooccur CTCF_ENCFF660GHM.bed RAD21_ENCFF057JFH.bed hg38_gene_hancer_v4.4.bed output.tsv
2022-01-20 01:24:40 [INFO] Calculate the co-occurrence of two sets of genomic intervals ...
2022-01-20 01:24:40 [INFO] Read and union BED file: "CTCF_ENCFF660GHM.bed"
2022-01-20 01:24:41 [INFO] Read and union BED file: "RAD21_ENCFF057JFH.bed"
2022-01-20 01:24:41 [INFO] Read and union background BED file: "hg38_gene_hancer_v4.4.bed"
2022-01-20 01:24:42 [INFO] Build interval tree for : "CTCF_ENCFF660GHM.bed"
2022-01-20 01:24:42 [INFO] Build interval tree for: "RAD21_ENCFF057JFH.bed"
A.name CTCF_ENCFF660GHM.bed
B.name RAD21_ENCFF057JFH.bed
A.count 58584
B.count 31955
G.count 218099
A+,B- 11545
A-,B+ 2525
A+,B+ 19602
A-,B- 184427
odds-ratio 124.0137
p-value 0.0000
Name: Fisher's exact test result, dtype: object
- A.count
Number of unique genomic intervals in “CTCF_ENCFF660GHM.bed”.
- B.count
Number of unique genomic intervals in “RAD21_ENCFF057JFH.bed”.
- G.count
Number of unique genomic intervals in background “hg38_gene_hancer_v4.4.bed” (g).
- A+,B-
Number of unique genomic intervals that are overlapped with A not B (a).
- A-,B+
Number of unique genomic intervals that are overlapped with B not A (b).
- A+,B+
Number of unique genomic intervals that are overlapped with both A and B (c).
- A-,B-
Number of unique genomic intervals that are overlapped with neither A nor B (n).